Life Sciences companies that want to sell their products or services in the United States must comply with the 21 CFR Part 11, Electronic Records and Electronic Signatures regulation. This includes systems used in researching, manufacturing, and distributing product such as pharmaceuticals, medical devices, biological products (e.g. vaccines), blood, and tissue (e.g. organ transplants).
It is critical that the professionals who build and support the systems used by these companies understand Part 11 in order to help life science companies be compliant.
The History of 21 CFR Part 11
The FDA introduced the 21 CFR Part 11 regulation at the request of industry. In the early 1990’s, computerized systems were being widely used by FDA regulated industries. Companies wanted to reduce or eliminate the need for paper-based records and signatures, but first needed to know what the FDA required. So they asked the FDA to provide guidelines for the use of electronic records and signatures.
The infographic below provides a brief history of the Part 11 regulation.
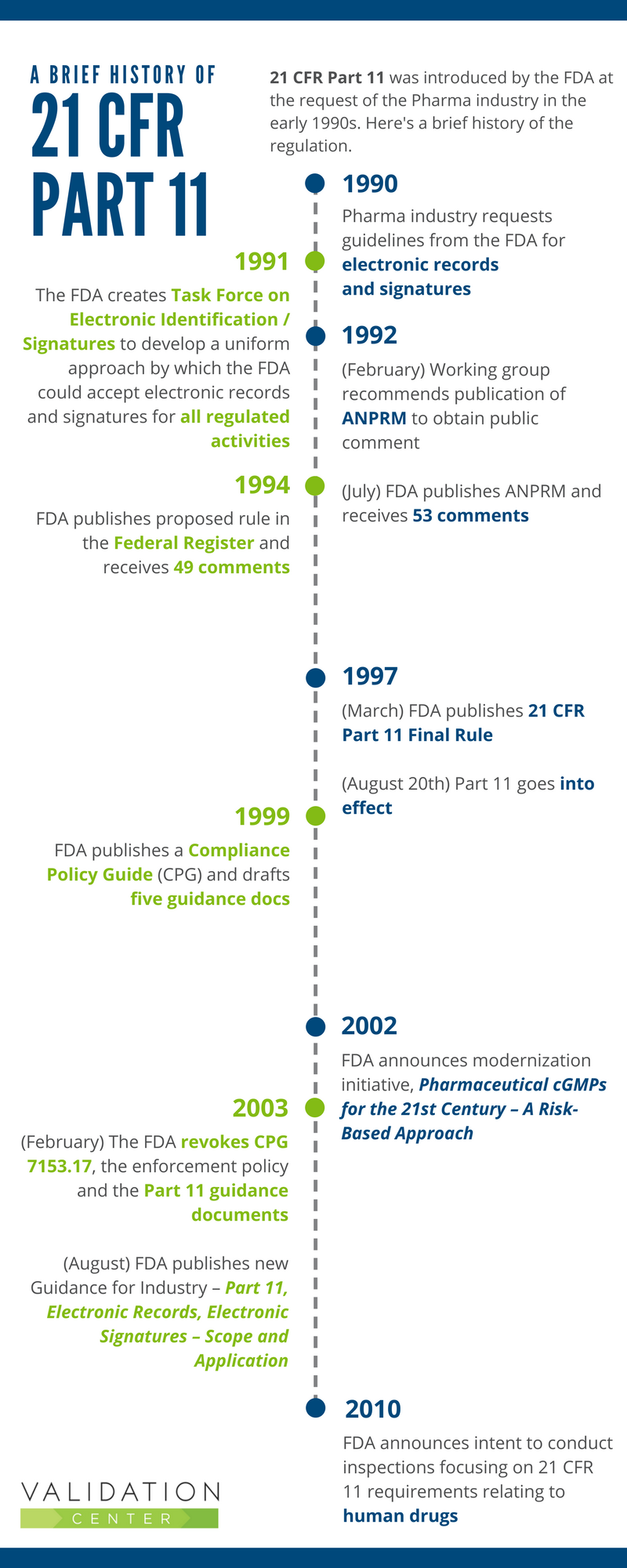
Part 11, The Early Years
In the late 1990s, when Part 11 went into effect, there was wide variation in the interpretation and implementation approaches used. Companies were unsure about which systems and records were within the scope of Part 11. They were also unclear on the specifics of how to implement the requirements. That led to the publication of the Compliance Policy Guide (CPG) and drafted Guidance Documents:
- CPG 7153.17: Enforcement Policy: 21 CFR Part 11; Electronic Records; Electronic Signatures
- Guidance: 21 CFR Part 11; Validation
- Guidance: 21 CFR Part 11; Glossary of Terms
- Guidance: 21 CFR Part 11; Time Stamps
- Guidance: 21 CFR Part 11; Maintenance of Electronic Records
- Guidance: 21 CFR Part 11; Electronic Copies of Electronic Records
In spite of the FDA’s efforts to provide clear, consistent approaches to Part 11 compliance, FDA regulated industries continued to express significant concerns regarding the regulation. The industry felt that 21 CFR Part 11 compliance:
- Restricted the use of technology (it seemed easier to stay on paper)
- Significantly increased the cost of using technology (systems needed customization to comply)
- Discouraged innovation and technological advances (because of cost and fear of non-compliance)
- Failed to provide significant benefit to public health
During this same time period, the FDA was encouraging manufacturing companies to adopt Process Analytical Technologies (PAT) to monitor processes in-line and on-line to enhance process understanding, consistency, and efficiency.
However, uncertainty around Part 11 compliance requirements was impeding companies from embracing PAT’s beneficial, data-centric technologies.
Modernization of Part 11
The 2002 publication Pharmaceutical cGMPs for the 21st Century – A Risk-Based Approach, was created to encourage adoption of new technological advances and implement risk-based approaches.
The following year, the industry was stunned to learn that the FDA had revoked the CPG 7153.17: Enforcement Policy as well as the five guidance documents. However, the 21 CFR Part 11 regulation is still in effect.
In August 2003, when the FDA published a new guidance for industry Part 11, Electronic Records; Electronic Signatures – Scope and Application, the key messages were:
- Part 11 remains in effect
- Part 11 will be re-examined
- The FDA will narrowly interpret the scope of Part 11 and apply enforcement discretion during this re-examination period.
We care about the Part 11 regulation because it is the FDA’s standard for considering an electronic record or signature to be trustworthy and reliable. It establishes minimums for the electronic equivalence to paper records and handwritten signatures.
The bottom line is, if you want to get away from pen and paper, you need to comply with Part 11 for activities regulated by the FDA.
We should pursue Part 11, not just because it’s a regulation
The FDA lists the benefits of Part 11 below:
- Protection and retrieval of electronic records
- Operational consistency
- Improve productivity and efficiency through automation
- Minimize or eliminate management of paper documentation
- Enable faster data-related searches
- Enable trending
- Electronic submissions to the FDA
Everyone can benefit from Part 11, starting with the FDA. Part 11 ensures the inspectability of electronic records. It makes sure the electronic records are available for the defined retention period, just like paper records. Additionally, organizations who move to electronic systems benefit from standardization, productivity, and efficiencies.
Part 11 also benefits the public because companies with electronic records can provide better analyses of quality data trends. Additionally, electronic submissions for FDA approval of new products are faster than paper submissions, meaning we should see products on the market sooner.
Need more help achieving Part 11 compliance?
If you are new to Part 11, make sure you have a solid understanding of the regulation. Attend our free 90-minute Fundamentals of Part 11 webinar or register for our Introductory training course.
Do you have significant gaps in your programs or need to prepare for an upcoming inspection? Our expert consultants can help you quickly implement an inspection-ready program or validate your systems.