
6 Steps to Prepare for an FDA Inspection (Don’t Leave it to Chance)
By Deb Bartel,
This post was recently updated on September 19, 2018
The FDA called. They’re coming to do an inspection. Yikes! Now what? How do you prepare?
We’ve been there. We’ve experienced the FDA Investigator’s arrival, unannounced, ready to inspect our computer systems.
It’s intimidating to say the least, especially if this is your first audit. The repercussions of a failed audit can be devastating to your organization. Warning letters are scary. Bad audits can be detrimental to the future of your organization.
So while you may not know when an investigator will knock on your door, this post will give you a practical approach to prepare for an FDA inspection.
Why would the FDA inspect your software system?
If the FDA is inspecting your system they are trying to determine if your system is compliant with government requirements. These audits are part of the FDA’s routine.
If you have a new product launching or a major product change of an existing product, the FDA will audit you prior to approval. Expect the same thing if you are opening a new facility or introducing your product to a new market, say on another continent. Other times the FDA would perform an inspection could be due to customer complaints or adverse events.
Follow these six steps to prepare for an FDA inspection
Step 1 – Review Audit Standards
The first step is to review audit standards, in this case FDA regulations. You will need to do your homework to find the applicable standards. 21 CFR Part 11 is a common audit standard for FDA-regulated companies. It contains all of the technical and procedures required for electronic records and electronic signatures.
Part 11 Guidance further explains which systems are in scope for Part 11 and the General Principles of Software Validation addresses the FDA’s expectations regarding how to validate medical device, manufacturing, quality management and other software used for FDA-regulated activities.
Audit Standards for Pharmaceutical Organizations
Pharma companies should read through 21 CFR 210 and 21 CFR 211. These audit standards contain the requirements for manufacturing, processing, packing or holding of drugs, and finished pharmaceuticals. Additionally, read this 1983 FDA Guide to Inspection of Computerized Systems in Drug Processing. It is old, but FDA investigators have referenced this document’s content during an audit.
Audit Standards for Biologics Manufacturers
If your company is manufacturing biological products, such as flu vaccines, you’ll want to review 21 CFR 600. This regulation contains supplemental requirements for your industry.
Audit Standards for Medical Device Manufacturers
Med Device manufacturers need to be familiar with 21 CFR 820. This Quality System Regulation contains basic requirements. Additionally, you might need to reference some guidance documents that go into details, such as risk assessment and management, change control, virus protection, and networks:
- Off-The-Shelf Software Use in Medical Devices
- Contents for Premarket Submissions for Software Contained in Medical Devices
Audit Standards for Blood or Blood Component Manufacturers
If your company is manufacturing blood or blood components, such as plasma, you’ll want to read 21 CFR 606. This regulation contains supplemental requirements for your industry. It also refers you back to the drug and device CFRs for basic requirements.
Audit Standards for working with Clinical Study Organizations
If you are working through a clinical study business, look at 21 CFR 50 and 21 CFR 56. Although these regulations don’t specifically cover computers, they do specify records you need to keep and how long to retain them.
Then read the guidance document, Computerized Systems Used in Clinical Investigations. This is a great guidance document, because it’s very logical, easy to read, and directly applicable. It also contains a nice list in the appendix of what SOPs you should have.
Audit Standards for Non-Clinical Laboratories
And finally, if you’re in a non-clinical lab, check out 21 CFR 58. Like the clinical regulation, this one doesn’t specifically address computers, but it does include computer data in the definition of ‘raw data’, and throughout the document gives many requirements for dealing with raw data.
Step 2 – Identify Which Items are in the Audit Scope
After you comb through the regulations and guidance documents relevant to your company, identify which systems will be part of the audit.
Identify systems that have processes supported by computer systems
For example, if your audit will cover a manufactured product where computers control the manufacturing equipment, then manufacturing control software is in scope. In another example, laboratory testing is in scope if computers run the lab instruments. Therefore, lab instrument software is in scope.
Find systems that have records created or retained by computer systems
For example, the regulations require a defect correction and prevention program. A computer system manages the program, therefore the CAPA system is in scope.
Let’s look at another example. You keep required patient visit records on electronic forms. Your forms system is in scope. Other systems that could contain records supported by computer systems include document management systems for SOPs and manufacturing instructions, complaints systems, adverse event systems and change control systems.
Look in the regulations for specific computer system requirements
Additionally, when identifying systems that are in scope, look in the regulations for specific computer system requirements, like software validation and electronic signatures, or records and data requirements like records/data protection and records/data retention.
Step 3 – Develop Practice Questions
Use the Audit Standards you identified in the previous step to develop practice questions.
Example for Pharma Companies
CFR 211 requires that laboratory records contain “complete data derived from all tests necessary to assure compliance with established specifications” and that this includes “the initials or signature of the person who performs each test and the date(s) the tests were performed.”
Practice questions could include:
- Does the lab record system include the initials or signature of the person who performed each test?
- Has the lab record system been validated?
- Is the validation documentation available?
- Does the validation documentation demonstrate the required feature?
- Does the electronic signature documentation meet the requirements of Part 11? (e.g., audit trail, human readable, readily retrievable, secure…)
Example for Medical Devices
21 CFR 820 requires that complaint records include device name, complaint date, any device ID numbers, complainant info <name, address, phone>, description of problem, date and results of investigation, etc. CFR 820 also requires that you retain records for the life of the device, but no less than 2 years after distribution.
So, practice questions could include:
- Do the complaint system records include all of the required data?
- Has the complaint system been validated?
- Is the validation documentation available?
- Does the validation documentation demonstrate the required feature?
- Are the system’s records retained for the required duration?
- Are there approved procedures for using and maintaining the system?
- Are there records showing that system users and support personnel were trained?
For more practice audit questions, attend our free 90-minute webinar.
Step 4 – Staff Your Auditee Team
An important step in the preparation process is to identify who will be on the auditee team, as well as their roles. You will need people who know the systems and IT processes. Team members can include key users, technical support, validation, and user support.
Determine who will perform the following:
- Speak to each topic during the audit
- Lead the facility tour
- Present the overview during the opening meeting
- Escort the auditors during the audit
- Take minutes of the audit
Typically, but not always, the Audit Facilitator performs items 2-5.
Another thing to consider will be the size of the audit. If your audit is going to be large, with many auditors, consider appointing a ‘back room coordinator’. This person contacts auditee team members when an auditor requests information from their designated area. The coordinator also screens the documents before sending them into the audit room.
Once you identify the members of the auditee team, provide audit interface training.
Step 5 – Prepare Your Documentation for the Inspection
To prepare for the audit you need to locate and review your documentation. This is where the auditee team has the chance to get familiar with existing documentation. We often find that documentation created long ago has not been reviewed in a while.
For each system in scope, find and review:
- Validation documentation
- Change control documentation
- Design documentation
- Incident documentation
- Maintenance documentation
- Back-up and recovery documentation
- Training plans and training materials
- SOPs – System Support (IT) SOPs, User SOPs
Track down lost or misplaced documentation
Make sure the documents are available and in good physical shape. Tape and fresh staples can work wonders.
Tip: Photocopy paper documentation that has deteriorated and should no longer be handled. Make a note as to where you can locate the originals during the audit.
Organize your documents in a way that allows you to quickly locate them
You never want to keep an inspector waiting while you dig through piles of paperwork. You look unprepared and careless. The inspector might decide to add more items to their inspection list! At the very least, they will scrutinize every detail.
If your documentation is not in the same location as the audit, you can:
- Bring copies of the documentation to the audit site
- Scan the documentation so that it’s available online
- Have a person on ‘stand-by’ to fax or email documents upon request – make sure this person is sitting by a phone during the audit
Understand previous documentation conventions
If your company has changed validation methodologies or software life cycles over the years, make sure you understand the old methodologies. If changes are big, make a document to cross-reference the deliverables.
Verify testing results support validation conclusion
When preparing your documentation for the audit, testing results should meet the predefined acceptance criteria and support the Validation Summary concluding that the system is suitable for use.
Ensure documentation signatures are not missing
It’s an easy thing to miss, but you can bet the inspectors will notice.
If documents are missing, signatures are missing, or any other problems detected during this step:
- Acknowledge the problem in writing (e.g., in CAPA, white paper, Remediation Plan)
- Identify the consequence of the problem
- Identify the course of action to remedy the problem
- Have management approve, in writing
Step 6 – Mock Audit
The last step of the audit preparation process is to hold a practice audit. Like rehearsing a speech, a mock audit will get the jitters out. Team members practice their roles, and you will get a sense for how the auditee team will handle the real inspection.
Someone who has been through many audits acts as the FDA Inspector, typically the Audit Facilitator. During the mock audit, ask questions covering areas with known issues. Request the evidence and ask the auditees to explain the documentation. The auditee team should take this mock audit seriously and respond as if the audit was real.
Other Pre-Inspection Activities
Just a few other areas indirectly related to the audit for you to review.
Update your org charts
Most auditors will request a copy and refer to it during the audit to see who they’re speaking with.
Make certain to follow security procedures
If you have a policy that everyone wears ID badges, then everyone needs to wear their ID badges. If you have a ‘secure’ facility, make sure locked doors are not propped open or deactivated.
Ensure facilities are neat, clean and in good repair
Look at your facilities through the eyes of a realtor. During the audit your facility is on display. Get rid of cobwebs in the corners, duct tape on cabinets and clutter in the file rooms. Other common no-no items include procedural violations such as password lists or cheat sheets.
How can you get it all done in time for the audit?
Perhaps you can’t. Especially when FDA Inspectors often show up unannounced. But, you can prioritize the work to get through as much as you can. If you don’t know where to begin prioritizing your audit preparation, use the following data to get started.
Start with systems that get the most citations
This 3-year summary shows which systems the FDA has been citing in warning letters most often:
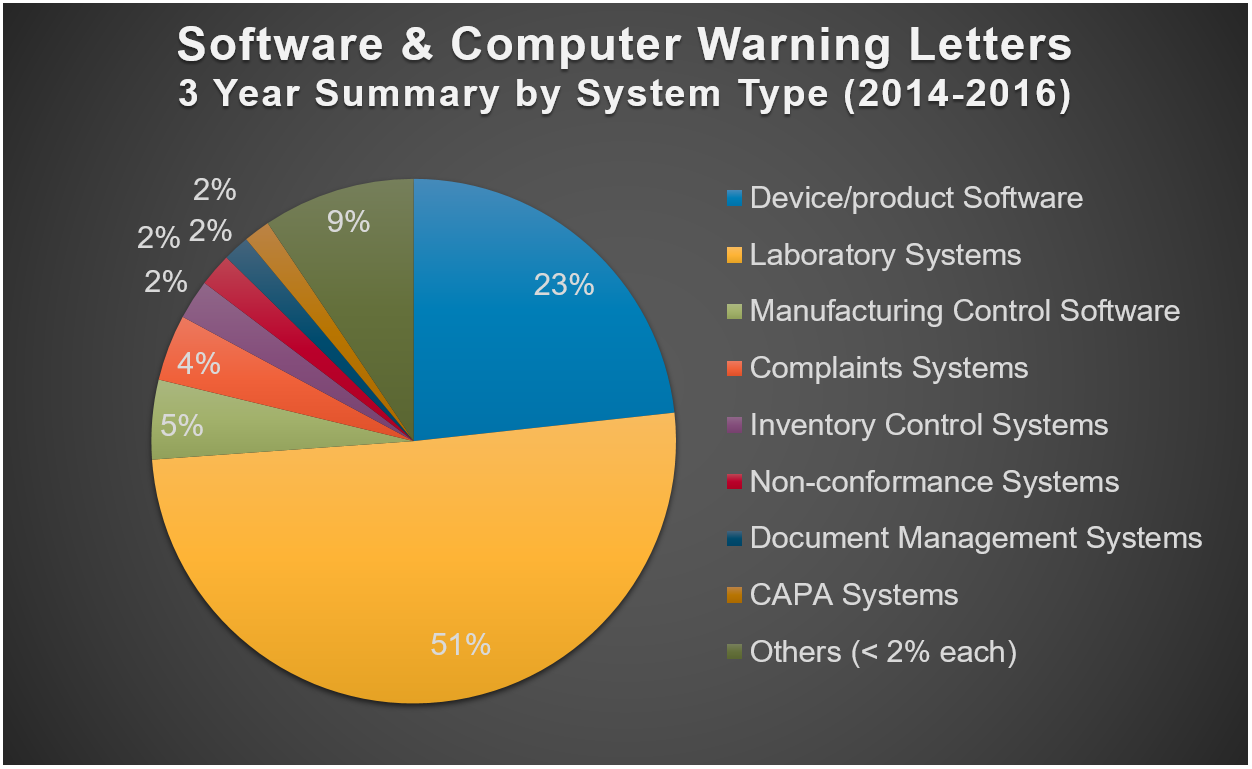
How should you interpret this information? Refer to the highest-cited systems as your number one priority. For example, if you have a lab system and a training system that both need validation but you can only do one at a time, start with the lab system.
The largest percentage of all software and computer system observations are on validation
This graph shows the relative citation frequency for software and computer system topics cited in recent FDA warning letters.

You can potentially avoid problems simply by doing validation
Over half (53%) of the validation observations in the chart above were for either:
- Failure to validate the system, or
- Failure to validate changes to the system
The remaining 47% of validation observations were for specific deficiencies in the validation process, such as inadequate validation SOPs, insufficient testing, no testing/unfinished testing and inadequate test evidence.
As in the system type example, focus your efforts on the biggest citation areas as you work your way through the preparation process.
If you are facing an FDA inspection, it’s not too late to get help.
Attend our free 90-minute Preparing for a Software Quality Audit webinar.
Have a pro at your side. Our veteran, certified audit facilitators can be on site with you, or coaching from behind the scenes.
Copyright Policy
Unless otherwise noted, Praxis Life Sciences, LLC is the legal copyright holder of all (written, multimedia and graphic) material on this website and it may not be used, reprinted, (partially) modified or published without written consent. A link to Validation Center must appear in all copies of any artwork or content, including articles and press releases.
You are welcome to share this article on social media so long as you link back directly to this post (providing the link). No copy & paste of this content will be allowed under any circumstances.
